Re-assembly
When a putative genome bin has been extracted it can often improve the assembly, i.e. reduce the number of scaffolds, if the reads associated with the bin are reassembled.
The rationale behind reassembly is two things. First of all the full metagenome was assembled with general parameters that wasn’t optimised for any particular genome. Hence, the assembly can be optimised with e.g. a kmer value that better fits the target genome. Secondly, re-assembling a subset of reads allows much better control of parameters that will limit the infulence of micro-diversity on the assembly.
Extract all reads associated with a bin
To reassemble a putative genome bin, we first need to extract the associated reads. The script extract.fasta.from.sam.using.list.pl takes a list of scaffolds l (e.g. as the genome1.txt file exported in the Binning section) and extracts all reads associated with these scaffolds from a SAM file s. The SAM file is the mapping of all metagenome reads to all assembled scaffolds (see PE tracking). The output o is a fasta file with the reads that were mapped to the supplied scaffolds.
perl multi-metagenome/reassembly/extract.fasta.from.sam.using.list.pl -l list.of.scaffolds.txt -s mapping.sam -o reads.subset.fa
Using this approach we only get the reads that already mapped to the scaffolds, i.e. Read1 in the figure below. However, we can use the mapped Read1 to extract the associated Read2. This is important if we want to improve the assembly.
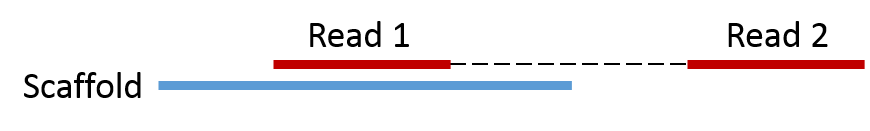
The script extract.fasta.pe.reads.using.single.pl takes the reads.subset.fa (s) that was extracted from the SAM file and extracts all associated read pairs in the original paried reads p (note: requires the paired reads in interleaved fasta format). The output is two fasta files p1.fa and p2.fa, which is read1 and read2 respectively.
perl multi-metagenome/reassembly/extract.fasta.pe.reads.using.single.pl -s reads.subset.fa -p all.paired.reads.fa
Reassembly of the extracted reads
As all reads associated with the bin of interest have been extracted, they can now be reassembled using any standard genome assembler. For reassembly we often use velvet as it gives more flexibility in tuning the assembly parameters compared to CLC and also since CLC currently only support a maximum kmer of 63. To reduce the impact of micro-diversity we normally try to assemble with a kmer as high as possible, e.g. a kmer of 99. The use of a large kmer does two things. First of all it increases the uniqeness of the kmers, which allows assembly of more closely related DNA regions. Secondly, it lowers the effective coverage of your dataset, whereby some of the low-abundant micro-diversity might get below the treshholds for interfering with the assembly.
Note that you need to optimise all assembly parameters independently for each kmer as different kmer values changes the (kmer)-coverage.
Automatic gap closing
Many assemblers leave gaps (N’s) in the scaffolds as they do not use a post-assembly gap filling module. We have recently started using the small tool GapFiller, which seem to work nicely.
Other assembly tweaks
If your target genome is in high coverage and have low abundant micro-diversity that interfers with the assembly an easy improvement is simply to use less data. Removing reads below X abundance can also be done using the khmer software.